A variant calling pipeline to analyse sequencing Illumina data

Project description



This is is the variant_calling pipeline from the Sequana projet
- Overview:
Variant calling from FASTQ files
- Input:
FASTQ files from Illumina Sequencing instrument
- Output:
VCF and HTML files
- Status:
production
- Citation:
Cokelaer et al, (2017), ‘Sequana’: a Set of Snakemake NGS pipelines, Journal of Open Source Software, 2(16), 352, JOSS DOI https://doi:10.21105/joss.00352
Installation
You must install Sequana first (use –upgrade to get the latest version installed):
pip install sequana --upgrade
Then, just install this package:
pip install sequana_variant_calling --upgrade
Usage
sequana_variant_calling --help sequana_variant_calling --input-directory DATAPATH --reference-file measles.fa sequana_variant_calling --input-directory DATAPATH --reference-file measles.fa
This creates a directory variant_calling. You just need to execute the pipeline:
cd variant_calling sh variant_calling.sh
This launch a snakemake pipeline. If you are familiar with snakemake, you can retrieve the pipeline itself and its configuration files and then execute the pipeline yourself with specific parameters:
snakemake -s variant_calling.rules -c config.yaml --cores 4 --stats stats.txt
Or use sequanix interface.
Requirements
This pipelines requires the following executable(s):
bwa
freebayes
picard (picard-tools)
sambamba
samtools
snpEff
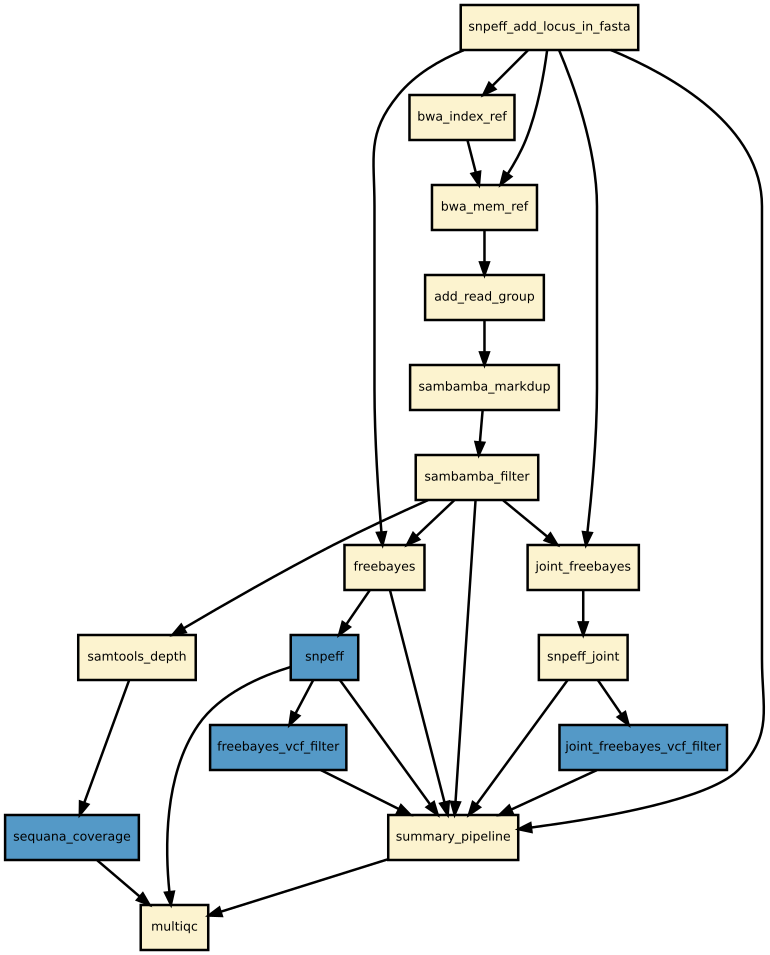
Details
Snakemake variant calling pipeline is based on tutorial written by Erik Garrison. Input reads (paired or single) are mapped using bwa and sorted with sambamba-sort. PCR duplicates are marked with sambamba-markdup. Freebayes is used to detect SNPs and short INDELs. The INDEL realignment and base quality recalibration are not necessary with Freebayes. For more information, please refer to a post by Brad Chapman on minimal BAM preprocessing methods.
The pipeline provides an analysis of the mapping coverage using sequana coverage. It detects and characterises automatically low and high genome coverage regions.
Detected variants are annotated with SnpEff if a GenBank file is provided. The pipeline does the database building automatically. Although most of the species should be handled automatically, some special cases such as particular codon table will required edition of the snpeff configuration file.
Finally, joint calling is also available and can be switch on if desired.
Changelog
Version |
Description |
|---|---|
0.10.0 |
|
0.9.10 |
|
0.9.5 |
|
0.9.4 |
|
0.9.3 |
|
0.9.2 |
|
0.9.1 |
|
0.9.0 |
First release |
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
File details
Details for the file sequana_variant_calling-0.10.0.tar.gz.
File metadata
- Download URL: sequana_variant_calling-0.10.0.tar.gz
- Upload date:
- Size: 340.0 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/2.0.0 pkginfo/1.5.0.1 requests/2.22.0 setuptools/41.6.0 requests-toolbelt/0.9.1 tqdm/4.50.2 CPython/3.8.9
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
5824f9a3d8685c9527affaf1a8f0bcf538d2a19b7238d972fcec573dea939fe1
|
|
| MD5 |
a37c328f754278033883d60182ba3aeb
|
|
| BLAKE2b-256 |
969716c238324bcb3028380622a46c639237e17b56083f5a174c15fe83565b84
|







